Sometimes when reading the residence time literature, I get the impression that the off-raters have re-animated Maxwell's Demon. It seems as if a nano-doorman stands guard at at the entrance of the binding site, only opening his nano-door to ligand molecules that want to get in. Microscopic Reversibility? Stop being so negative! With Big Data, Artificial Intelligence, Machine Learning (formerly known as QSAR) and Ligand Efficiency Metrics we can beat Microscopic Reversibility and consign The Second Law to the Dustbin Of History!
There were a number of things that triggered this blog post. First, I saw a recent article that got me thinking about philatelic drug discovery. Second, some of the off-raters will be getting together in Berlin next week and I wanted to share some musings because I won't be there in person. Third, my former colleague Rutger Folmer has published a useful (dare I say, brave) critique of the residence time concept that is bang on target.
I'm not actually going to say much about Rutger's article except to suggest that you read it. That's because I really want to examine the article on philatelic drug discovery in a more detail (it's actually about thermodynamic and kinetic profiling but I thought the reference to philately would better grab your attention). My standard opening move when playing chess with an off-rater is to assert that slow binding is equivalent to slow distribution. In what situations would you design a drug to distribute slowly?
Chemical kinetics is all about energy barriers and, the higher the barrier, the slower things will happen. Microscopic reversibility tells us that a barrier to association is a barrier to dissociation and that the ligand will return to solution along the same path that it took to its binding site. Microscopic reversibility tells you that if you got into the parking spot you can get out of it as well although that may not be the experience of every driver. The reason that microscopic reversibility doesn't always seem to apply to parking is that most humans, with the possible exception of tank drivers in the Italian army, are more comfortable in forward gear than in reverse. Molecules, in contrast, have no more concept of forward and reverse than they do of standard states, IUPAC or the opinions the 'experts' who might quantitatively estimate their drug-likeness while judging their beauty. Molecules don't actually do concepts. Put more uncouthly, molecules just don't give a toss.
I've created a graphic to illustrate to show how things might look in vivo when there is a barrier to association (and, therefore, to dissociation). We can think of the ligand molecule having to get over the barrier in order to get to its binding site and we call the top of the barrier the 'transition state'. This is a simplified version of reality (it is actually the system that passes from the unbound state through the transition state to the bound state and for some ligand-protein association there is no barrier) but it'll serve for what I'd like to say. The graphic consists of three panels and the first (A) of these illustrates the situation soon after dosing when the concentration of ligand (L) is relatively high and the target protein (P) has not had sufficient time to respond. If the barrier is sufficiently high, the system can't get to equilibrium before the ligand concentration starts to fall in what a pharmacokineticist might refer to as the elimination phase. Under this scenario the system will be at equilibrium briefly as the ligand concentration falls and I've shown this in panel B. After the equilibrium point is reached, the rate of dissociation exceeds the rate of association and this is shown in panel C.

There's something else that I'd like you to take a look at in the graphic and that's the free energy (G) of the unbound state (P + L). See how it goes down relative to the free energy of the bound state (P.L) as the concentration of ligand decreases. When thinking about energetics of these systems, it actually makes a lot of sense to use the unbound state as the reference but you do need to use a reference concentration (e.g. 1 M) to to do this.
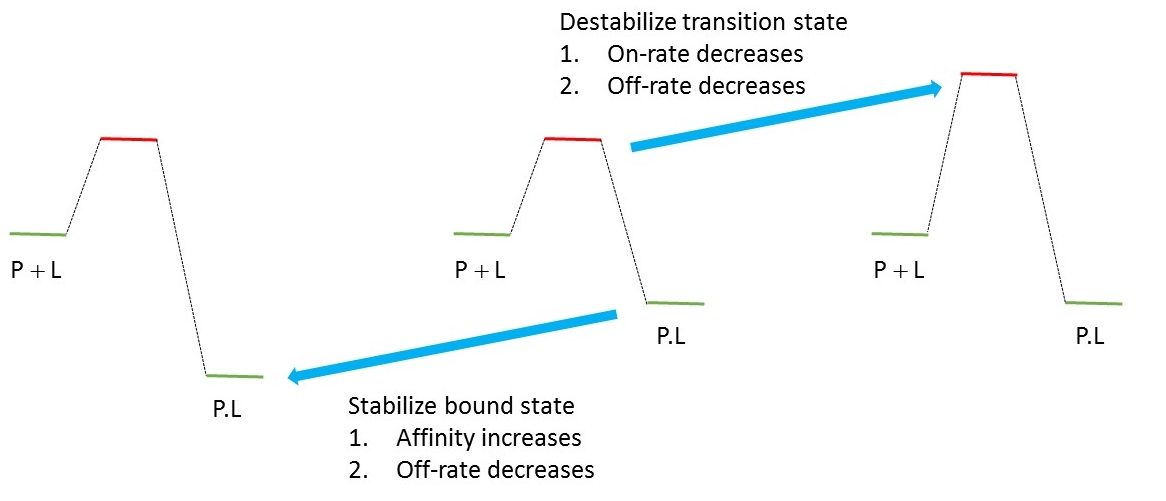
When we do molecular design we often think in terms of manipulating energy differences. For example, we try to increase affinity by stabilizing the bound state relative to the unbound state. Once you start trying to manipulate off-rates, you soon realize that you can't change one thing at a time (unless you draft Maxwell's Demon into your project team). I've created a second graphic which looks similar to the first graphic although there are important differences between the two graphics. In particular, I'm referencing energy to the unbound state (P + L) which means that the ligand concentration is constant in all three panels. Let's consider the central panel as the starting point for design. We can go left from that starting point and stabilize the bound state which is equivalent to optimizing affinity. Stabilizing the bound state will also result in slower dissociation provided that the transition stare energy remains unchanged. This is a good thing but it's difficult to show that the benefits come from the slower dissociation and not from the increased affinity. If you raise the barrier (i.e. increase the energy of the transition state) to reduce the off-rate you'll find that you have slowed the on-rate to an equal extent.

Before moving on, it may be useful to sum up where we've got to so far. First, ask yourself why you think off-rates will be relevant in situations where concentration changes on a longer time scale than binding. Second, you'll need to enlist the help of Maxwell's Demon if you want to reduce off-rate without affecting on-rate and/or affinity. Third, if you want to consider binding kinetics in design then it'd be best to use barrier height (referenced to unbound state) and affinity as your design parameters.
Now I'd take a look at the philatelic drug discovery article. This is a harsh term but it does capture a tendency in some drug discovery programs to measure things for the sake of it (or at least to keep the grinning Lean Six Sigma 'belts' grinning). Some of this is a result of using techniques such as isothermal titration calorimetry (ITC) and surface plasmon resonance (SPR) that yield information in addition to affinity (that is of primary interest) at no extra cost. I really don't want to come across as a Luddite and I must stress that measurements of enthalpy, entropy, on-rate and off-rate are of considerable scientific interest and are also valuable for improving physical models. Furthermore, I am continually awed by the exquisite sensitivity of modern ITC and SPR instruments and would always want the option to be able to measure affinity using at least one of these techniques. However, problems start when the access to enthalpy, entropy, off-rates and on-rates becomes exploited for 'metrication' and drug discovery scientists seek 'enthalpy-driven' binding simply because the binding will be more 'enthalpy-driven'. It is easier to make the case for relevance of binding kinetics although, as Rutger points out, reducing the off-rate may very well make things worse if the on-rate is also reduced. It is much more difficult to assemble a coherent case for the relevance of thermodynamic signatures in drug discovery. Perhaps, some day, a seminal paper from the Budapest Enthalpomics Group (BEG) will reveal that isothermal systems like live humans can indeed sense the enthalpy and entropy changes associated with drugs binding to their targets although I will not be holding my breath.
Unsurprisingly, the thermodynamic and kinetic profiling (aka philatelic drug discovery) article advocates thermodyanamic profiling of bioactive compounds in lead optimization projects. I'm going to focus on the kinetic profiling and it is worrying that the authors don't seem to be aware that on-rates and off-rates have to be seen in a pharmacokinetic context in order to make the connection with drug discovery. The authors may find it instructive to think about how inhibitor concentration would have varied over the course of a typical experiment in their cell-based assays. They are also likely to find Rutger's article to be educational and I recommend that they familiarize themselves with its content.
The following statement suggests that it may be beneficial for the authors to also familiarize themselves with the rudiments of chemical kinetics:
Some analysis of relationships between potency in a cell-based assay and KD, koff and kon were presented in Figure 6 of the article. I have a number of gripes with the analysis. First, it would be better to use logarithms of quantities like KD, IC50, koff and kon when performing analysis of this nature. In part, this because we typically look for linear free energy relationships in these situations. There is another strong rationale for using logarithms because analysis of correlations between continuous variables works best when the uncertainties in data values are as constant as possible. My second gripe is that the authors have chosen to bin their data for analysis and this is a great way to shoot yourself in the foot. When you bin continuous data you both reduce your data analysis options and leave people wondering whether the binning has been done to hide the weakness of the trends in the data. I have droned at length about why it is naughty to bin continuous data so I'll leave it at that.
It's been a long post and it's time to wrap things up. If you've found the post to be 'cansativo' (sounds so much more soothing in Portguese) then spare a thought for the person who had to write it. To conclude, I'll leave you with a quote that I've taken from the abstract for Rutger's article:
"Moreover, fast association is typically more desirable than slow, and advantages of long residence time, notably a potential disconnect between pharmacodynamics (PD) and pharmacokinetics (PK), would be partially or completely offset by slow on-rate."
Chemical kinetics is all about energy barriers and, the higher the barrier, the slower things will happen. Microscopic reversibility tells us that a barrier to association is a barrier to dissociation and that the ligand will return to solution along the same path that it took to its binding site. Microscopic reversibility tells you that if you got into the parking spot you can get out of it as well although that may not be the experience of every driver. The reason that microscopic reversibility doesn't always seem to apply to parking is that most humans, with the possible exception of tank drivers in the Italian army, are more comfortable in forward gear than in reverse. Molecules, in contrast, have no more concept of forward and reverse than they do of standard states, IUPAC or the opinions the 'experts' who might quantitatively estimate their drug-likeness while judging their beauty. Molecules don't actually do concepts. Put more uncouthly, molecules just don't give a toss.
I've created a graphic to illustrate to show how things might look in vivo when there is a barrier to association (and, therefore, to dissociation). We can think of the ligand molecule having to get over the barrier in order to get to its binding site and we call the top of the barrier the 'transition state'. This is a simplified version of reality (it is actually the system that passes from the unbound state through the transition state to the bound state and for some ligand-protein association there is no barrier) but it'll serve for what I'd like to say. The graphic consists of three panels and the first (A) of these illustrates the situation soon after dosing when the concentration of ligand (L) is relatively high and the target protein (P) has not had sufficient time to respond. If the barrier is sufficiently high, the system can't get to equilibrium before the ligand concentration starts to fall in what a pharmacokineticist might refer to as the elimination phase. Under this scenario the system will be at equilibrium briefly as the ligand concentration falls and I've shown this in panel B. After the equilibrium point is reached, the rate of dissociation exceeds the rate of association and this is shown in panel C.

There's something else that I'd like you to take a look at in the graphic and that's the free energy (G) of the unbound state (P + L). See how it goes down relative to the free energy of the bound state (P.L) as the concentration of ligand decreases. When thinking about energetics of these systems, it actually makes a lot of sense to use the unbound state as the reference but you do need to use a reference concentration (e.g. 1 M) to to do this.
When we do molecular design we often think in terms of manipulating energy differences. For example, we try to increase affinity by stabilizing the bound state relative to the unbound state. Once you start trying to manipulate off-rates, you soon realize that you can't change one thing at a time (unless you draft Maxwell's Demon into your project team). I've created a second graphic which looks similar to the first graphic although there are important differences between the two graphics. In particular, I'm referencing energy to the unbound state (P + L) which means that the ligand concentration is constant in all three panels. Let's consider the central panel as the starting point for design. We can go left from that starting point and stabilize the bound state which is equivalent to optimizing affinity. Stabilizing the bound state will also result in slower dissociation provided that the transition stare energy remains unchanged. This is a good thing but it's difficult to show that the benefits come from the slower dissociation and not from the increased affinity. If you raise the barrier (i.e. increase the energy of the transition state) to reduce the off-rate you'll find that you have slowed the on-rate to an equal extent.
Before moving on, it may be useful to sum up where we've got to so far. First, ask yourself why you think off-rates will be relevant in situations where concentration changes on a longer time scale than binding. Second, you'll need to enlist the help of Maxwell's Demon if you want to reduce off-rate without affecting on-rate and/or affinity. Third, if you want to consider binding kinetics in design then it'd be best to use barrier height (referenced to unbound state) and affinity as your design parameters.
Now I'd take a look at the philatelic drug discovery article. This is a harsh term but it does capture a tendency in some drug discovery programs to measure things for the sake of it (or at least to keep the grinning Lean Six Sigma 'belts' grinning). Some of this is a result of using techniques such as isothermal titration calorimetry (ITC) and surface plasmon resonance (SPR) that yield information in addition to affinity (that is of primary interest) at no extra cost. I really don't want to come across as a Luddite and I must stress that measurements of enthalpy, entropy, on-rate and off-rate are of considerable scientific interest and are also valuable for improving physical models. Furthermore, I am continually awed by the exquisite sensitivity of modern ITC and SPR instruments and would always want the option to be able to measure affinity using at least one of these techniques. However, problems start when the access to enthalpy, entropy, off-rates and on-rates becomes exploited for 'metrication' and drug discovery scientists seek 'enthalpy-driven' binding simply because the binding will be more 'enthalpy-driven'. It is easier to make the case for relevance of binding kinetics although, as Rutger points out, reducing the off-rate may very well make things worse if the on-rate is also reduced. It is much more difficult to assemble a coherent case for the relevance of thermodynamic signatures in drug discovery. Perhaps, some day, a seminal paper from the Budapest Enthalpomics Group (BEG) will reveal that isothermal systems like live humans can indeed sense the enthalpy and entropy changes associated with drugs binding to their targets although I will not be holding my breath.
Unsurprisingly, the thermodynamic and kinetic profiling (aka philatelic drug discovery) article advocates thermodyanamic profiling of bioactive compounds in lead optimization projects. I'm going to focus on the kinetic profiling and it is worrying that the authors don't seem to be aware that on-rates and off-rates have to be seen in a pharmacokinetic context in order to make the connection with drug discovery. The authors may find it instructive to think about how inhibitor concentration would have varied over the course of a typical experiment in their cell-based assays. They are also likely to find Rutger's article to be educational and I recommend that they familiarize themselves with its content.
The following statement suggests that it may be beneficial for the authors to also familiarize themselves with the rudiments of chemical kinetics:
"Association
and dissociation rate constants (kon and koff) of compound binding to a
biological target are not intrinsically related to one another, although they
are connected by dissociation equilibrium constant KD (KD = koff/kon)."
The processes of association and dissociation are actually connected by virtue of taking place along the same path and by having to pass through the same transition states. The difference in barrier heights for association and dissociation is given by the binding free energy. Some analysis of relationships between potency in a cell-based assay and KD, koff and kon were presented in Figure 6 of the article. I have a number of gripes with the analysis. First, it would be better to use logarithms of quantities like KD, IC50, koff and kon when performing analysis of this nature. In part, this because we typically look for linear free energy relationships in these situations. There is another strong rationale for using logarithms because analysis of correlations between continuous variables works best when the uncertainties in data values are as constant as possible. My second gripe is that the authors have chosen to bin their data for analysis and this is a great way to shoot yourself in the foot. When you bin continuous data you both reduce your data analysis options and leave people wondering whether the binning has been done to hide the weakness of the trends in the data. I have droned at length about why it is naughty to bin continuous data so I'll leave it at that.
It's been a long post and it's time to wrap things up. If you've found the post to be 'cansativo' (sounds so much more soothing in Portguese) then spare a thought for the person who had to write it. To conclude, I'll leave you with a quote that I've taken from the abstract for Rutger's article:
"Moreover, fast association is typically more desirable than slow, and advantages of long residence time, notably a potential disconnect between pharmacodynamics (PD) and pharmacokinetics (PK), would be partially or completely offset by slow on-rate."