<< previous | next >>
I last posted on Covid-19 early in 2021 and quite a lot has happened since then. Specifically, a number of vaccines are now available (I received my first dose of AstraZeneca CoviShield in May 2021 while still stranded in Trinidad) and paxlovid has been approved for use as a Covid-19 treatment (Derek describes his experiences taking paxlovid in this post). The active ingredient of paxlovid is the SARS-CoV-2 main protease inhibitor nirmatrelvir and the ritonavir with which it is dosed serves only to reduce clearance of nirmatrelvir by inhibiting metabolic enzymes. In the current post, I’ll be looking at covalent inhibition of SARS-CoV-2 main protease with a specific focus on reversibility and here are some notes that I whipped up as a contribution to the Covid Moonshot.
Nirmatrelvir (1) is shown in Chart 1 along with SARS-CoV-2 main protease inhibitors from the Covid Moonshot (2), a group of (mainly) Sweden-based academic researchers (3) and Yale University (4). Nirmatrelvir incorporates a nitrile group that forms a covalent bond with the catalytic cysteine and the other inhibitors bind non-covalently to the target. The first example of a nitrile-based cysteine protease inhibitor that I’m aware of was published over half a century ago and the nitrile warhead has since proved popular with designers of cysteine protease inhibitors (it has a small steric footprint and is not generally associated with metabolic lability or chemical instability). Furthermore, covalent bond formation between the thiol of a catalytic cysteine and the carbon of the nitrile warhead is typically reversible. Here’s a recent review on the nitrile group in covalent inhibitor design and this comparative study of electrophilic warheads may also be of interest.

At this point, we should be thinking about the directions in which design of SARS-CoV-2 main protease inhibitors needs to go. Two directions I see as potentially productive are dose reduction (a course of paxlovid treatment consists of two 150 mg nirmatrelvir tablets and one 100 mg ritonavir tablet taken twice daily for five days) and countering resistance (here’s a relevant article).
Two tactics for achieving a lower therapeutic dose are to increase affinity and reduce clearance. Dose prediction is not as easy as you might think because the predictions are typically very sensitive to input parameters. For example, a two-fold difference in IC50 would often be regarded as within normal assay variation by medicinal chemists but development scientists and clinicians would view doses of 300 mg and 600 mg very differently.
Excessive clearance is a problem from the perspective of achieving adequate exposure and I'd also anticipate greater variability in exposure between patients when clearance is high. Clearance is clearly an issue for nirmatrelvir because it needs be co-dosed with ritonavir (to inhibit metabolic enzymes) and this has implications for patients taking other medications. Nirmatrelvir lacks aromatic rings and deuteration is an obvious tactic to reduce metabolic lability (although cost of goods is likely to be more of an issue than for a cancer medicine that you'll need to take out a second mortgage for). I would anticipate that bicyclo[1.1.1]pentanyl will be less prone to metabolism than t-butyl (CH bonds tend to be stronger in strained rings and for bridgehead CHs) and the binding mode suggests that this replacement could be accommodated.
Details of resistance to nirmatrelvir (P2022 | Z2022) are starting to emerge and this information should be certainly be used in design and to assess other structural series. Nevertheless, if you’re genuinely concerned about potential for resistance then you really can’t afford to ignore conserved structural features in the target such as the catalytic residues (cysteine and histidine) and the oxyanion hole. I would also anticipate that the risk of resistance will increase with the spatial extent of the inhibitor.
This post is about covalent inhibitors. Although I’m pleasantly surprised by the potencies achieved for non-covalent SARS-Cov-2 Main Protease inhibitors, I consider making a virtue of non-covalent inhibition to be a serious error. Binding of covalent inhibitors to their targets can be reversible or irreversible and, in the context of design, reversible covalent inhibitors have a lot more in common with non-covalent inhibitors than with irreversible covalent inhibitors (for example, you can't generally use mass spectroscopy to screen covalent fragments that bind reversibly). In the context of drug design, covalent bonds have much more stringent geometric requirements than non-covalent interactions such as hydrogen bonds.
I generally favor reversible binding when targeting catalytic cysteines as discussed in these notes and this article. It is typically less difficult to design reversible covalent inhibitors to target a catalytic cysteine than it is to design irreversible covalent inhibitors because you can use crystal structures of protein-ligand complexes just as you would for non-covalent inhibitors. In contrast, the crystal of a protein-ligand complex (the reaction ‘product’) is not especially relevant in design of irreversible inhibitors because target engagement is under kinetic rather than thermodynamic control and the more relevant transition state models must therefore be generated computationally. Furthermore, assays for irreversible inhibitors are more complex, and assessment of functional selectivity and safety is more difficult than for reversible inhibitors. All that said, however, I’m certainly not of the view that irreversible inhibitors are inherently inferior to reversible inhibitors for targeting catalytic cysteines. This is also a good point to mention an article which shows how isosteric replacement (with an alkyne) of the nitrile warhead of the reversible cathepsin K inhibitor odanacatib results in an irreversible inhibitor (the article is particularly relevant if you’re interested in chemical probes for cysteine proteases).
I contributed some designs for reversible covalent inhibitors to the Covid Moonshot and it may be helpful to discuss some of them. Each design was intended to link the nitrile warhead to the ‘3-aminopyridine-like’ scaffold used in the Covid Moonshot which means that the designs all use a heteroaromatic P1 group (typically isoquinoline linked at C4) rather than the chiral P1 group (pyrrolidinone linked at C3) used for nirmatrelvir and a number of other SARS-CoV-2 main protease inhibitors. The ‘3-aminopyridine-like’ scaffold lacks essential hydrogen bond donors (elimination of hydrogen bond donors is suggested as a tactic for increasing aqueous solubility in this article). One of the cool things about the way the Covid Moonshot was set up is that I can link designs as they were originally submitted (often with a detailed rationale and proposed binding mode).
The most direct way to link a nitrile to the ‘3-aminopyridine-like’ scaffold is with methylene (5, Chart 2) but there is a problem with this approach because substituting anilides (and their aza-analogs) on nitrogen with sp3 carbon inverts the cis/trans geometrical preference of the anilides (I discussed the design implications of this in these notes). This implies that binding of 5 to the target is expected to incur a conformational energy penalty and it is significant that N-methylation of 6 results in a large reduction in potency. Although 5 was inactive in the enzyme inhibition assay, I think that it would still be worth seeing if covalent bond formation can be observed by crystallography for this compound.

However, you won’t invert cis/trans geometrical preference if you substitute an anilide nitrogen with nitrogen rather than sp3 carbon (Chart 3). This was the basis for submitting 8, which is related to azapeptide nitriles, as a design. Azapeptide nitriles [L2008 | Y2012 | L2019 | B2022] are typically more potent than the corresponding peptide nitriles and, to be honest, this remains something of a mystery to me (one possibility is that the imine nitrogen of the azapeptide nitrile adduct is more basic than that of the corresponding peptide nitrile adduct and is predominantly protonated under assay conditions). I see cyanohydrazines and cyanamides as functional groups that would be worth representing in fragment libraries if you want to target catalytic cysteine residues and I’ll point you toward a relevant crystal structure. The acyclic hydrazine and cyanamide substructures in 8 trigger structural alerts although there are approved drugs that incorporate acyclic hydrazine (atazanavir | bumadizone | gliclazide | goserelin | isocarboxazid | isoniazid) and N-cyano (cimetidine) substructures. The basis for these structural alerts is obscure and it’s worth noting that 8 is incorrectly flagged as an enamine and having a nitrogen-oxygen single bond. As a cautionary tale on structural alerts, I’ll refer you to this comment in which I read the riot act (i.e., the JMC guidelines for authors) to a number of ACS journal EiCs Nevertheless, I’d still worry about the presence of an acyclic hydrazine substructure although these concerns would be eased if each nitrogen atom was bonded to an electron-withdrawing group, as is the case for 8, and all NHs were capped (see 9).

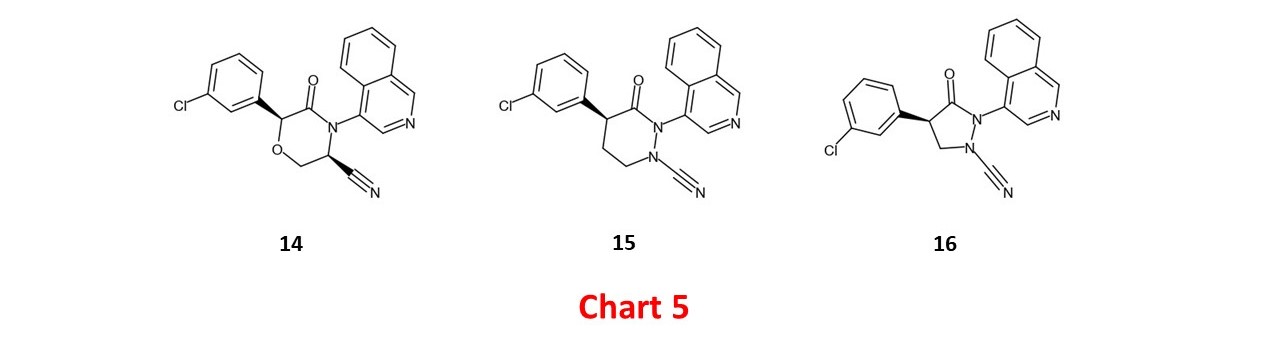
An alternative tactic to counter inversion of the cis/trans geometrical preference is to lock the conformation with a ring and designs 10 and 11 (Chart 4) can be seen as 'hybrids' of 5 with 12 and 13 respectively (in fragment-based design, hybridization is usually referred to as fragment merging). The effect of the conformational lock can be clearly seen since 12 and 13 are essentially equipotent with 6 (the primary reason for proposing 12 and 13 as designs was actually to present the nitrile warhead to the catalytic cysteine). A substituent on carbon next to a lactam nitrogen tends to adopt an axial orientation and I’d anticipate that 10 will be less prone to epimerization than 11. Although I'm unaware of nitriles being deployed on cyclic amine substructures for cysteine protease inhibition, the structures of the DPP-4 inhibitors saxagliptin and vildagliptin are relevant.
This is a good point at which to wrap up. If cysteine protease inhibition is a key component of pandemic preparedness strategy then you really do need to be thinking about covalent inhibition. I'll be looking at some more design themes for covalent inhibitors of SARS-CoV-2 in the next Covid post.