A couple of months ago I enjoyed a visit to the US (my first for eight years) on which I caught up with old friends before and after a few days in Vermont (where a trip to the golf course can rapidly become a National Geographic Moment). One highlight of the trip was randomly meeting my friend and fellow blogger Ash Jogalekar for the first time in real life (we’ve actually known each other for about fifteen years) on the Boston T Red Line. Following a couple of nights in green and leafy Belmont, I headed for the Flatlands with an old friend from my days in Minnesota for a Larry Miller group reunion outside Chicago before delivering a short harangue on polarity at Ripon College in Wisconsin. After the harangues, we enjoyed a number of most excellent Spotted Cattle (Only in Wisconsin) in Ripon. I discovered later that one of my Instagram friends is originally from nearby Green Lake and had taken classes at Ripon College while in high school. It is indeed a small world.

The five days spent discussing computer-aided drug design (CADD) in Vermont are what I’ll be covering in this post and I think it’s worth saying something about what drugs need to do in order to function safely. First, drugs need to have significant effects on therapeutic targets without having significant effects on anti-targets such as hERG or CYPs and, given the interest in new modalities, I’ll be say “effects” rather than “affinity”, although Paul Ehrlich would have reminded us that drugs need to bind in order to exert effects. Second, drugs need to get to their targets at sufficiently high concentrations for their effects to be therapeutically significant (drug discovery scientists use the term ‘exposure’ when discussing drug concentration). Although it is sometimes believed that successful drugs simply reduce the numbers of patients suffering from symptoms it has been known from the days of Paracelsus that it is actually the dose that differentiates a drug from a poison.
Drug design is often said to be multi-objective in nature although the objectives are perhaps not as numerous as many believe (this point is discussed in the introduction section of NoLE, an article that I'd recommend to insomniacs everywhere). The first objective of drug design can be stated in terms of minimization of the concentration at which a therapeutically useful effect on the target is observed (this is typically the easiest objective to define since drug design is typically directed at specific targets). The second objective of drug design can be stated in analogous terms as maximization of the concentration at which toxic effects on the anti-targets are observed (this is a more difficult objective to define because we generally know less about the anti-targets than about the targets). The third objective of drug design is to achieve controllability of exposure (this is typically the most difficult objective to define because drug concentration is a dose-dependent, spaciotemporal quantity and intracellular concentration cannot generally be measured for drugs in vivo). Drug discovery scientists, especially those with backgrounds in computational chemistry and cheminformatics, don’t always appreciate the importance of controlling exposure and the uncertainty in intracellular concentration always makes for a good stock question for speakers and panels of experts.
I posted previously on artificial intelligence (AI) in drug design and I think it’s worth highlighting a couple of common misconceptions. The first misconception is that we just need to collect enough data and the drugs will magically condense out of the data cloud that has been generated (this belief appears to have a number of adherents in Silicon Valley). The second misconception is that drug design is merely an exercise in prediction when it should really be seen in a Design of Experiments framework. It’s also worth noting that genuinely categorical data are rare in drug design and my view is that many (most?) "global" machine learning (ML) models are actually ensembles of local models (this heretical view was expressed in a 2009 article and we were making the point that what appears to be an interpolation may actually be an extrapolation). Increasingly, ML is becoming seen as a panacea and it’s worth asking why quantitative structure activity relationship (QSAR) approaches never really made much of a splash in drug discovery.
I enjoyed catching up with old friends [ D | K | S | R/J | P/M ] as well as making some new ones [ G | B/R | L ]. However, I was disappointed that my beloved Onkel Hugo was not in attendance (I continue to be inspired by Onkel’s laser-like focus on the hydrogen bonding of the ester) and I hope that Onkel has finally forgiven me for asking (in 2008) if Austria was in Bavaria. There were many young people at the gathering in Vermont and their enthusiasm made me greatly optimistic for the future of CADD (I’m getting to the age at which it’s a relief not to be greeted with: "How nice to see you, I thought you were dead!"). Lots of energy at the posters (I learned from one that Voronoi was Ukrainian) although, if we’d been in Moscow, I’d have declined the refreshments and asked for a room on the ground floor (left photo below). Nevertheless, the bed that folded into the wall (centre and right photos below) provided plenty of potential for hotel room misadventure without the ‘helping hands’ of NKVD personnel.

It'd been four years since CADD had been discussed at this level in Vermont so it was no surprise to see COVID-19 on the agenda. The COVID-19 pandemic led to some very interesting developments including the Covid Moonshot (a very different way of doing drug discovery and one I was happy to contribute to during my 19 month sojourn in Trinidad) and, more tangibly, Nirmatrelvir (an antiviral medicine that has been used to treat COVID-19 infections since early 2022). Looking at the molecular structure of Nirmatrelvir you might have mistaken trifluoroacetyl for a protecting group but it’s actually a important feature (it appears to be beneficial from the permeability perspective). My view is that the alkane/water logP (alkane is a better model than octanol for the hydrocarbon core of a lipid bilayer) for a trifluoroacetamide is likely to be a couple of log units greater than for the corresponding acetamide.
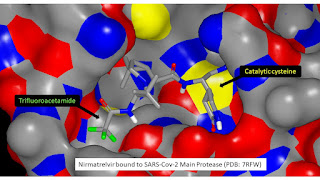
I’ll take you through how the alkane/water logP difference between a trifluoroacetamide and corresponding acetamide can be estimated in some detail because I think this has some relevance to using AI in drug discovery (I tend to approach pKa prediction in an analogous manner). Rather than trying to build an ML model for making the prediction, I’ve simply made connections between measurements for three different physicochemical properties (alkane/water logP, hydrogen bond basicity and hydrogen bond acidity) which is something that could easily be accommodated within an AI framework. I should stress that this approach can only be used because it is a difference in alkane/water logP (as opposed to absolute values) that is being predicted and these physicochemical properties can plausibly be linked to substructures.
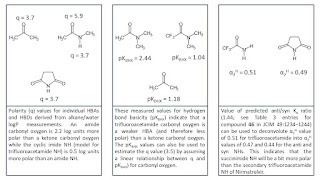
Let’s take a look at the triptych below which I admit that is not quite up to the standards of Hieronymus Bosch (although I hope that you find it to be a little less disturbing). The first panel shows values of polarity (q) for some hydrogen bond acceptors and donors (you can find these in Tables 2 and 3 in
K2022) that have been derived from alkane/water logP measurements. You could, for example, use these polarity values to predict that reducing the polarity of an amide carbonyl oxygen to the extent that it looks like a ketone will lead to a 2.2 log unit increase in alkane/water logP. The second panel shows measured hydrogen bond basicity values for three hydrogen bond acceptors (you can find these in this freely available
dataset) and the values indicate that a trifluoroacetamide is an even weaker hydrogen bond acceptor than a ketone. Assuming a linear relationship between polarity and hydrogen bond basicity, we can estimate that the trifluoroacetamide carbonyl oxygen is 2.4 log units less polar than the corresponding acetamide. The final panel shows measured hydrogen bond acidity values (you can find these in Table 1 of
K2022) that suggest that an imide NH (q = 1.3; 0.5 log units more polar than typical amide NH) will be slightly more polar than the trifluoroacetamide NH of Nirmatrelvir. So to estimate he difference in alkane/water logP values you just need to subtract the additional polarity of trifluoroacetamide NH (0.5 log units) from the lower polarity of the trifluoroacetamide carbonyl oxygen (2.4) to get 1.9 log units.
Chemical space is a recurring theme in drug design and its vastness, which defies human comprehension, has inspired much navel-gazing over the years (it’s actually tangible chemical space that’s relevant to drug design). In drug discovery we need to be able to navigate chemical space (ideally without having to ingest huge quantities of Spice) and, given that Ukrainian chemists have revolutionized the world's idea of
tangible chemical space (and have also made it a whole lot larger), it is most appropriate to have a Ukrainian
guide who is most ably assisted by a trusty Transylvanian sidekick. I see benefits from considering molecular complexity more explicitly when mapping chemical space.
AI (as its evangelists keep telling us) is quite simply awesome at generating novel molecular structures although, as noted in a previous
post, there’s a little bit more to drug design than simply generating novel molecular structures. Once you’ve generated a novel molecular structure you need to decide whether or not to synthesize the compound and, in AI-based drug design, molecular structures are often assessed using ML models for biological activity as well as absorption, distribution, metabolism and excretion (ADME) behaviour. It’s well-known that you need a lot of data for training these ML models but you also need to check that the compounds for which you’re making predictions lie within the chemical space occupied by the training set (one way to do this is to ensure that close structural analogs of these compounds exist in the training set) because you can’t be sure that the big data necessarily cover the regions of chemical space of interest to drug designers using the models. A panel discusses the pressing requirement for more data although ML modellers do need to be aware that there’s a huge difference between assembling data sets for
benchmarking and covering chemical space at sufficiently high resolution to enable accurate prediction for arbitrary compounds.
There are other ways to think about chemical space. For example, differences in biological activity and ADME-related properties can also be seen in terms of structural relationships between compounds. These structural relationships can be defined in terms of molecular similarity (Tanimoto coefficient for the molecular fingerprints of X and Y is 0.9) or substructure (X is the 3-chloro analog of Y). Many medicinal chemists think about structure-activity relationships (SARs) and structure-property relationships (SPRs) in terms of matched molecular pairs (MMPs: pairs of molecular structures that are linked by specific substructural relationships) and free energy perturbation (FEP) can also be seen in this framework. Strong
nonadditivity and
activity cliffs (large differences in activity observed for close structural analogs) are of considerable interest as SAR features in their own right and because prediction is so challenging (and therefore very useful for testing ML and physics-based models for biological activity). One reason that drug designers need to be aware of activity cliffs and nonadditivity in their project data is that these SAR features can potentially be exploited for selectivity.
Cheminformatic approaches can also help you to decide how to synthesize the compounds that you (or your AI Overlords) have designed and automated synthetic route planning is a prerequisite for doing drug discovery in ‘self-driving’ laboratories. The key to success in cheminformatics is getting your data properly organized before starting analysis and the Open Reaction Database (
ORD), an open-access schema and infrastructure for structuring and sharing organic reaction data, facilitates training of models. One area that I find very exciting is the use of high-throughput experimentation in the search for
new synthetic reactions which can led to better coverage of unexplored chemical space. It’s well known in industry that the process chemists typically synthesize compounds by routes that differ from those used by the medicinal chemists and data-driven multi-objective
optimization of catalysts can lead to more efficient manufacturing processes (a higher conversion to the desired product also makes for a cleaner crude product).
It’s now time to wrap up what’s been a long post. Some of what is referred to as AI appears to already be useful in drug discovery (especially in the early stages) although non-AI computational inputs will continue to be significant for the foreseeable future. I see a need for cheminformatic thinking in drug discovery to shift from big data (global ML models) to focused data (generate project specific data efficiently for building local ML models) and also see advantages in using atom-based descriptors that are clearly linked to molecular interactions. One issue for data-driven approaches to prediction of biological activity such as ML and QSAR modelling is that the need for predictive capability is greatest when there's not much relevant data and this is a scenario under which physics-based approaches have an advantage. In my view, validation of ML models is not a solved problem since clustering in chemical space can cause validation procedures to make optimistic assessments of model quality. I continue to have significant concerns about how relationships (which are not necessarily linear) between descriptors are handled in ML modelling and remain generally skeptical of claims for interpretability of ML models (as noted in
NoLE, the contribution of a protein–ligand contact to affinity is not, in general, an experimental observable).
Many thanks for staying with me to the end and hope to see many of you at
EuroQSAR in Barcelona next year. I'll leave you with a memory from the early days of chemical space navigation.




No comments:
Post a Comment